







2023

Alexander Mühleip, Rasmus Kock Flygaard, Rozbeh Baradaran, Outi Haapanen, Thomas Gruhl, Victor Tobiasson, Amandine Maréchal, Vivek Sharma & Alexey Amunts
Structural basis of mitochondrial membrane bending by the I–II–III2–IV2 supercomplex
Nature, 615, 934–938
Abstract | PDF Article
2022

Yuzuru Itoh, Vivek Singh, Anas Khawaja, Andreas Naschberger, Minh Duc Nguyen, Joanna Rorbach, Alexey Amunts
Structure of the mitoribosomal small subunit with streptomycin reveals Fe-S clusters and physiological molecules
eLife,11, e77460
Abstract | PDF Article
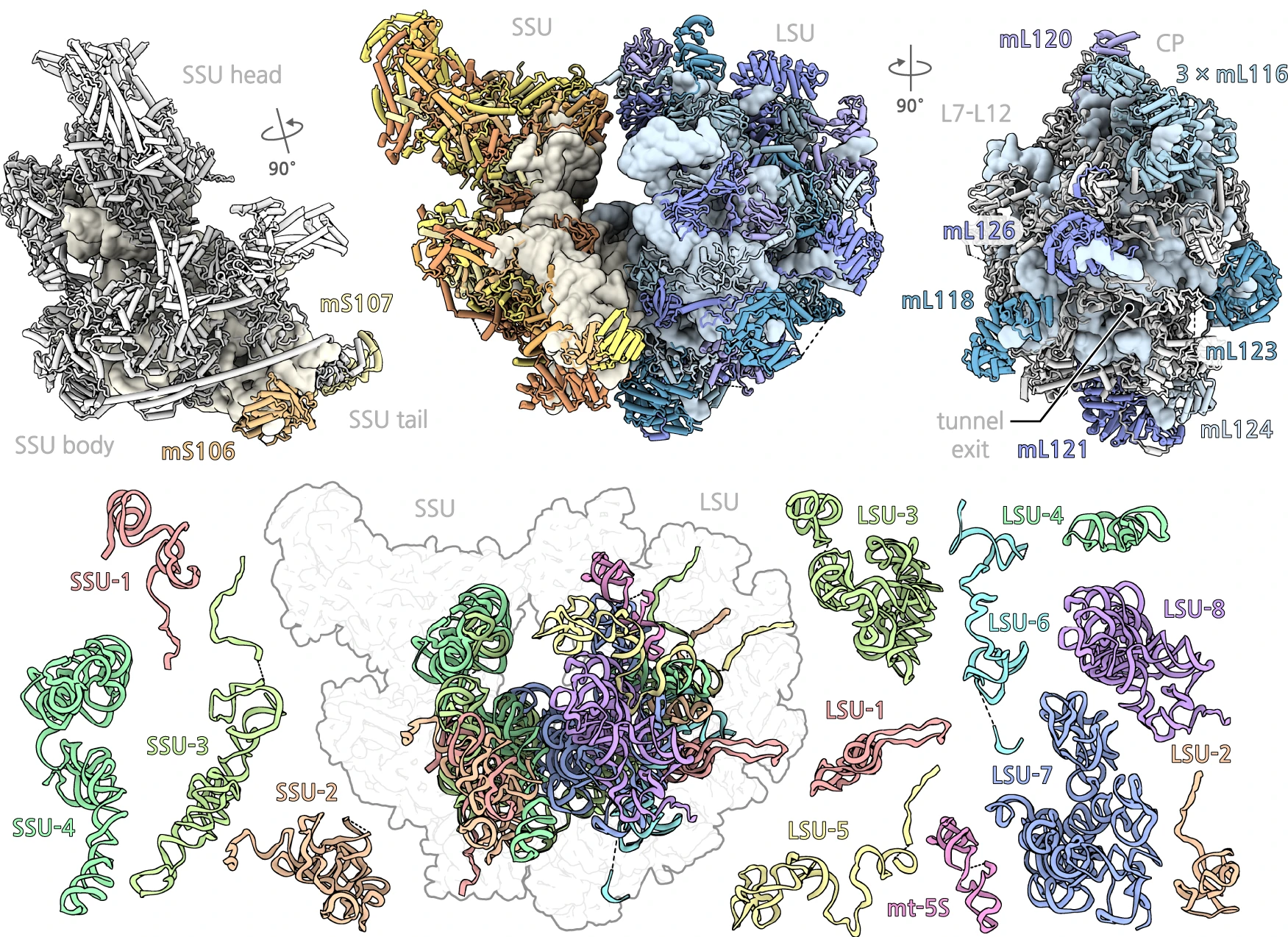
Victor Tobiasson, Ieva Berzina, Alexey Amunts
Structure of a mitochondrial ribosome with fragmented rRNA in complex with membrane-targeting elements
Nature Communications, 13, 6132
Abstract | PDF Article
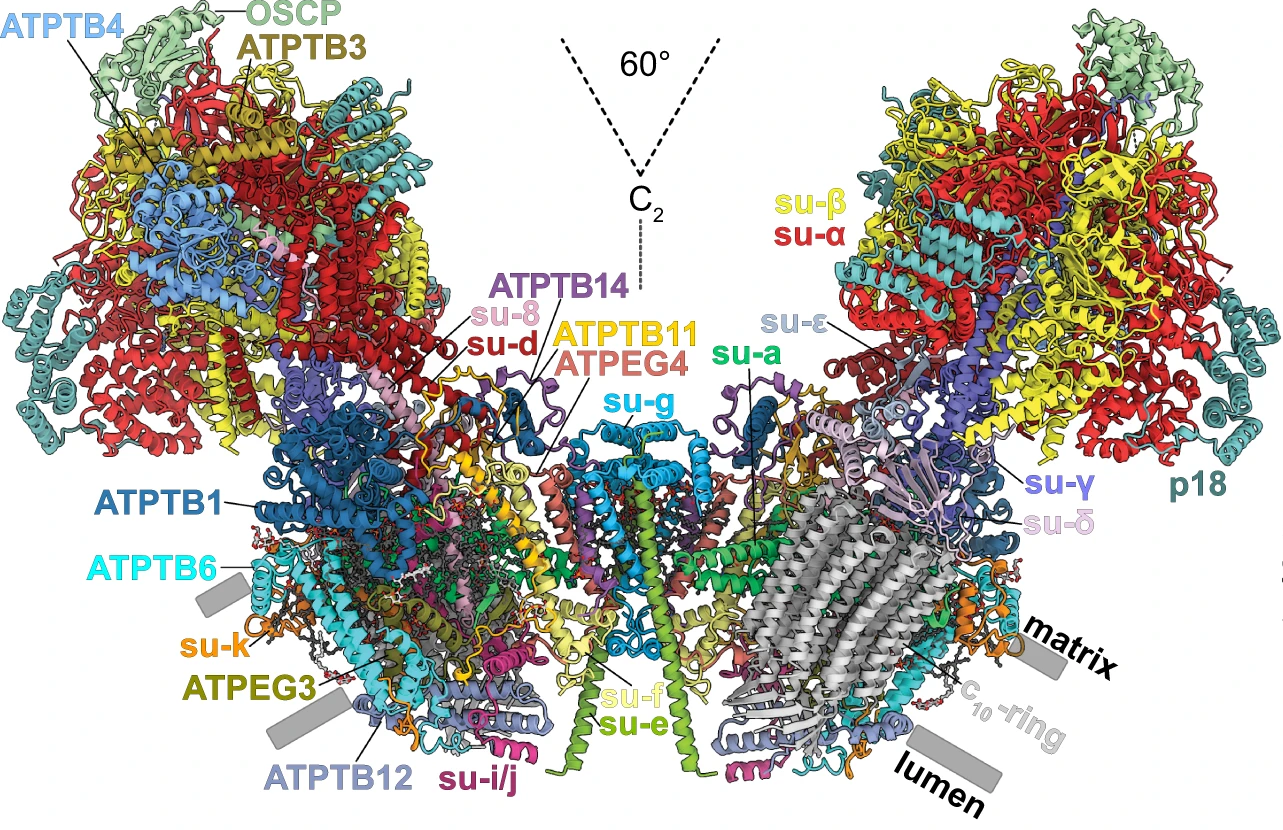
Ondřej Gahura, Alexander Mühleip, Carolina Hierro-Yap, Brian Panicucci, Minal Jain, David Hollaus, Martina Slapničková, Alena Zíková, Alexey Amunts
An ancestral interaction module promotes oligomerization in divergent mitochondrial ATP synthases
Nature communications, 13, 1-13
Abstract | PDF Article
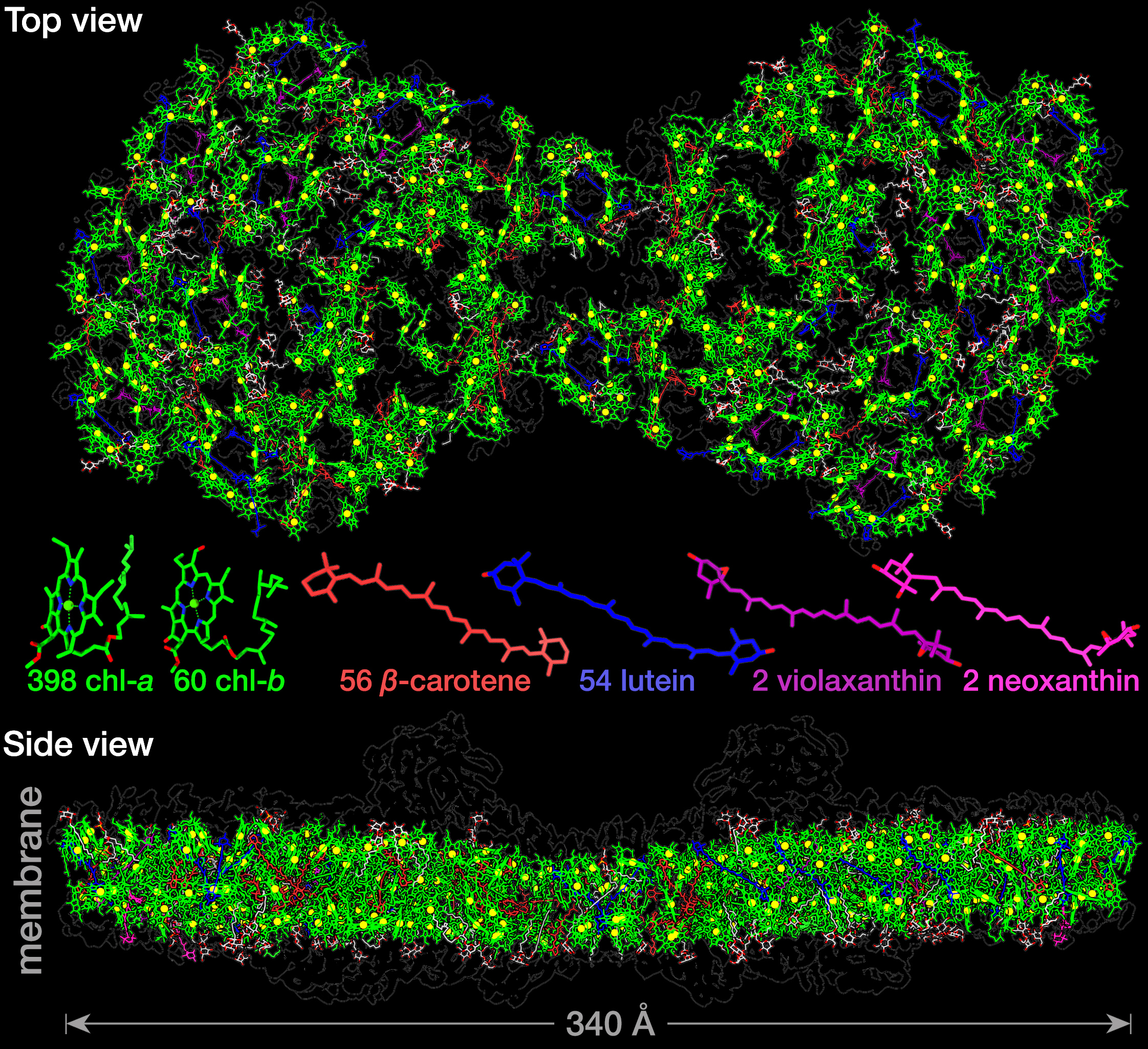
Andreas Naschberger, Laura Mosebach, Victor Tobiasson, Sebastian Kuhlgert, Martin Scholz, Annemarie Perez-Boerema, Thi Thu Hoai Ho, André Vidal-Meireles, Yuichiro Takahashi, Michael Hippler, Alexey Amunts
Algal photosystem I dimer and high-resolution model of PSI-plastocyanin complex
Nature Plants, 8, 1191–1201
Abstract | PDF Article
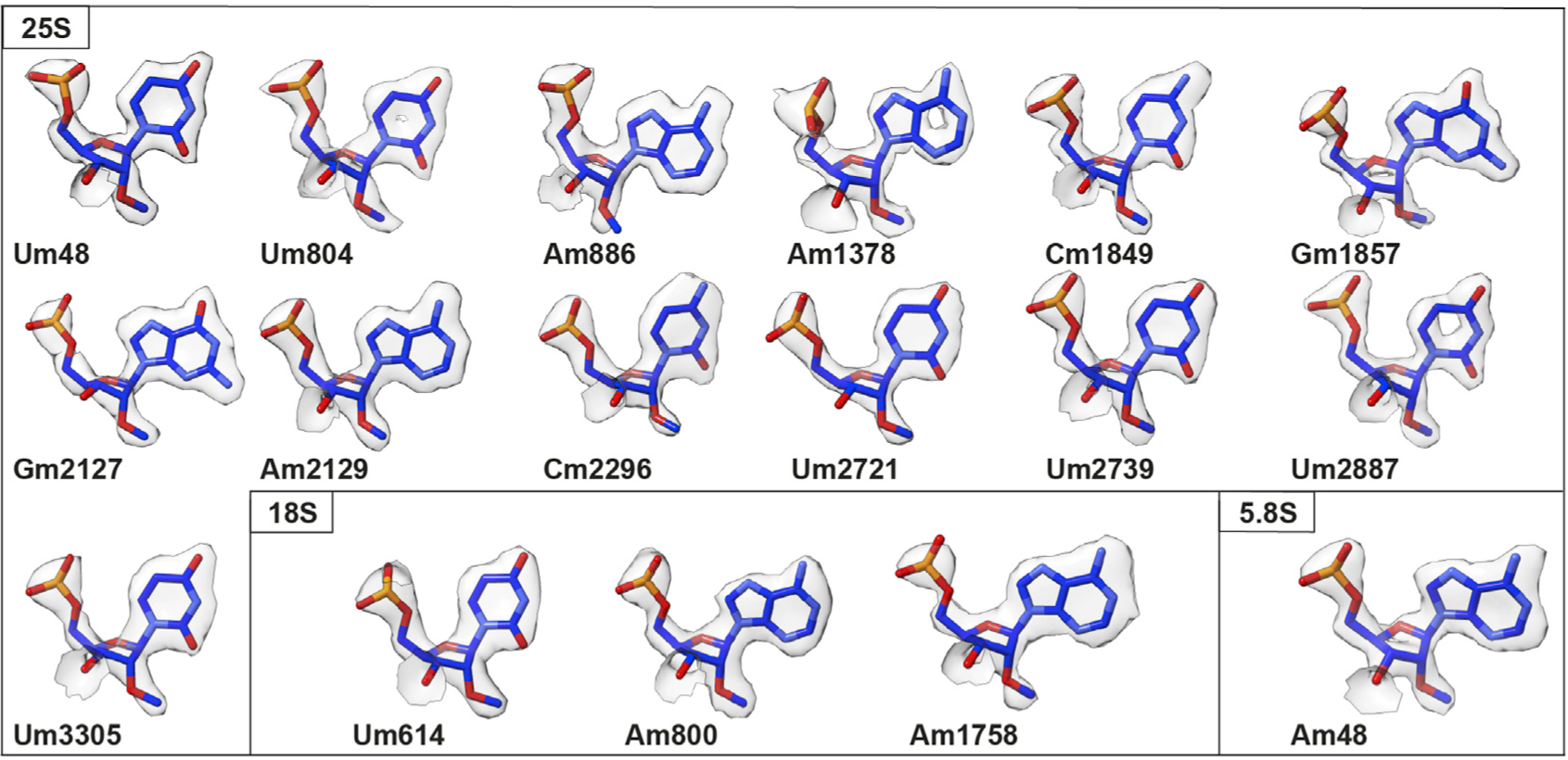
Patrick Cottilli, Yuzuru Itoh, Yuko Nobe, Anton Petrov, Puri Lisón, Masato Taoka, Alexey Amunts
Cryo-EM structure and rRNA modification sites of a plant ribosome
Plant Communications, 100342
Abstract | PDF Article

Yuzuru Itoh, Anas Khawaja, Ivan Laptev, Miriam Cipullo, Ilian Atanassov, Petr Sergiev, Joanna Rorbach, Alexey Amunts
Mechanism of mitoribosomal small subunit biogenesis and preinitiation
Nature, 606, 603–608
Abstract | PDF Article
2021

Yuzuru Itoh Y, Juni Andrell, Austin Choi, Uwe Richter, Priyanka Maiti, Robert Best, Antonio Barrientos, Brendan Battersby, Alexey Amunts
Mechanism of membrane-tethered mitochondrial protein synthesis
Science, 371, 846-849
Abstract | PDF Article

Victor Tobiasson, Ondrej Gahura, Shintaro Aibara, Rozbeh Baradaran, Alena Zíková, Alexey Amunts
Interconnected assembly factors regulate the biogenesis of mitoribosomal large subunit
The EMBO J, e106292
Abstract | PDF Article

Alexander Mühleip, Rasmus Kock Flygaard, Jana Ovciarikova, Alice Lacombe, Paula Fernandes, Lilach Sheiner, Alexey Amunts
ATP synthase hexamer assemblies shape cristae of Toxoplasma mitochondria
Nature Communications, 12, 120
Abstract | PDF Article
2020

Rasmus Kock Flygaard, Alexander Mühleip, Victor Tobiasson, Alexey Amunts
Type III ATP synthase is a symmetry-deviated dimer that induces membrane curvature through tetramerization
Nature Communications, 11, 5342
Abstract | PDF Article

Yuzuru Itoh, Andreas Naschberger, Narges Mortezaei, Johannes M. Herrmann, Alexey Amunts
Analysis of translating mitoribosome reveals functional characteristics of translation in mitochondria of fungi
Nature Communications, 11, 5187
Abstract | PDF Article

Shintaro Aibara, Vivek Singh, Angelika Modelska, Alexey Amunts
Structural basis of mitochondrial translation
eLife, 9 , e58362
Abstract | PDF Article

Victor Tobiasson, Alexey Amunts
Ciliate mitoribosome illuminates evolutionary steps of mitochondrial translation
eLife, 9 , e59264
Abstract | PDF Article

Anas Khawaja, Yuzuru Itoh, Cristina Remes, Henrik Spåhr, Olessya Yukhnovets, Henning Höfig, Alexey Amunts, Joanna Rorbach
Distinct pre-initiation steps in human mitochondrial translation
Nature Communications, 11 (1), pp. 1-10
Abstract | PDF Article

Ming Chen, Annemarie Perez-Boerema, Laixing Zhang, Yanxue Li, Maojun Yang, Shizhong Li, Alexey Amunts
Distinct structural modulation of photosystem I and lipid environment stabilizes its tetrameric assembly
Nature Plants, 6 (3), pp. 314-320
Abstract | PDF Article

Annemarie Perez-Boerema, Daniel Klaiman, Ido Caspy, Sigal Y Netzer-El, Alexey Amunts, Nathan Nelson
Structure of a minimal photosystem I from the green alga Dunaliella salina
Nature Plants, 6 (3), pp. 321-327
Abstract | PDF Article
2019

Alexander Mühleip, Sarah E McComas, Alexey Amunts
Structure of a mitochondrial ATP synthase with bound native cardiolipin
eLife, 8 , e51179
Abstract | PDF Article

E. Bugiardini, A. Mitchell, I. Rosa, H. Horning-Do, A. Pitmann, O. Poole, J. Holton, S. Shah, C. Woodward, I. Hargreaves, R. Quinlivan, A. Amunts , R. Wiesner, H. Houlden, I. Holt, M. Hanna, R. Pitceathly, A. Spinazzola
MRPS25 mutations impair mitochondrial translation and cause encephalomyopathy
Hum Mol Genet. 28(16): 2711-2719
Abstract | PDF Article

Neha Nirwan, Yuzuru Itoh, Pratima Singh, Sutirtha Bandyopadhyay, Kutti R. Vinothkumar, Alexey Amunts, Kayarat Saikrishnan
Structure-based mechanism for activation of the AAA+ GTPase McrB by the endonuclease McrC
Nature Communications, 10 (1), pp. 1-9
Abstract | PDF Article

Janna M Bigalke, Shintaro Aibara, Robert Roth, Göran Dahl, Euan Gordon, Sarah Dorbéus, Alexey Amunts, Jenny Sandmark
Cryo-EM structure of the activated RET signaling complex reveals the importance of its cysteine-rich domain
Science Advances, 5 (7), eaau4202
Abstract | PDF Article

Victor Tobiasson, Allexa Dow, Sladjana Prisic, Alexey Amunts
Zinc depletion does not necessarily induce ribosome hibernation in mycobacteria
Proceedings of the National Academy of Sciences, 116 (7), pp. 2395-2397
Abstract | PDF Article

Anton S Petrov, Elizabeth C Wood, Chad R Bernier, Ashlyn M Norris, Alan Brown, Alexey Amunts
Structural patching fosters divergence of mitochondrial ribosomes
Molecular biology and evolution, 36 (2), pp. 207-219
Abstract | PDF Article
2018

Shintaro Aibara, Juni Andréll, Vivek Singh, Alexey Amunts
Rapid isolation of the mitoribosome from HEK cells
JoVE (Journal of Visualized Experiments), 140 , pp. e57877
Abstract | PDF Article

Annemarie Perez Boerema, Shintaro Aibara, Bijoya Paul, Victor Tobiasson, Dari Kimanius, Björn O Forsberg, Karin Wallden, Erik Lindahl, Alexey Amunts
Structure of the chloroplast ribosome with chl-RRF and hibernation-promoting factor
Nature Plants, 4 (4), pp. 212-217
Abstract | PDF Article
2017

Joanna Rorbach, Shintaro Aibara, Alexey Amunts
Ribosome origami
Nature Structural & Molecular Biology, 24 (11), pp. 879-881
Abstract | PDF Article

Björn O. Forsberg, Shintaro Aibara, Dari Kimanius, Bijoya Paul, Erik Lindahl, Alexey Amunts
Cryo-EM reconstruction of the chlororibosome to 3.2 Å resolution within 24 h
IUCrJ, 4 (6), pp. 723-727
Abstract | PDF Article | Supplementary Information

Alan Brown, Sorbhi Rathore, Dari Kimanius, Shintaro Aibara, Xiao-chen Bai, Joanna Rorbach, Alexey Amunts, V. Ramakrishnan
Structures of the human mitochondrial ribosome in native states of assembly
Nature Structural & Molecular Biology, 24 (10), pp. 866–869
Abstract | PDF Article
Donna Matzov, Shintaro Aibara, Arnab Basu, Ella Zimmerman, Anat Bashan, Mee-Ngan F Yap, Alexey Amunts, Ada E Yonath
The cryo-EM Structure of Hibernating 100S Ribosome Dimer From Pathogenic Staphylococcus Aureus
Nature communications, 8 (1), pp. 1-7
Abstract | PDF Article

Nirupa Desai, Alan Brown, Alexey Amunts, V. Ramakrishnan
The structure of the yeast mitochondrial ribosome
Science, 355 (6324), pp. 528-531
Abstract | PDF Article
2016

Martin Ott, Alexey Amunts, Alan Brown
Organization and regulation of mitochondrial protein synthesis
Annual Review of Biochemistry, 85
Abstract | PDF Article
2015

Dasmanthie De Silva, Ya-Ting Tu, Alexey Amunts, Flavia Fontanesi, Antoni Barrientos
Mitochondrial ribosome assembly in health and disease
Cell Cycle, 14 (14), pp. 2226-2250
Abstract | PDF Article

Alexey Amunts, Alan Brown, Jaan Toots, Sjors HW Scheres, V. Ramakrishnan
The structure of the human mitochondrial ribosome
Science, 348 (6230), pp. 95-98
Abstract | PDF Article

Alexey Amunts, Karol Fiedorczuk, Thao T Truong, Josephine Chandler, E Peter Greenberg, V Ramakrishnan
Bactobolin A Binds to a Site on the 70S Ribosome Distinct from Previously Seen Antibiotics
Journal of Molecular Biology, 427 (4), pp. 753-755
Abstract | PDF Article
2014

Alan Brown, Alexey Amunts, Xiao-chen Bai, Yoichiro Sugimoto, Patricia C Edwards, Garib Murshudov, Sjors HW Scheres, V. Ramakrishnan
Structure of the large ribosomal subunit from human mitochondria
Science, 346 (6210), pp. 718-722
Abstract | PDF Article

Alexey Amunts, Alan Brown, Xiao-chen Bai, Jose L Llácer, Tanweer Hussain, Paul Emsley, Fei Long, Garib Murshudov, Sjors HW Scheres, Venki Ramakrishnan
Structure of the yeast mitochondrial large ribosomal subunit
Science, 343 (6178), pp. 1485-1489
Abstract | PDF Article
2011

Nicoleta Herascu, Mehdi Najafi, Alexey Amunts, Jörg Pieper, Klaus-Dieter Irrgang, Rafael Picorel, Michael Seibert, Valter Zazubovich
Parameters of the protein energy landscapes of several light-harvesting complexes probed via spectral hole growth kinetics measurements
The Journal of Physical Chemistry B, 115 (12), pp. 2737-2747
Abstract | PDF Article
2010

Alexey Amunts, Hila Toporik, Anna Borovikova, Nathan Nelson
Structure determination and improved model of plant photosystem I
Journal of Biological Chemistry, 285 (5), pp. 3478-3486
Abstract | PDF Article
2009

Alexey Amunts, Nathan Nelson
Plant photosystem I design in the light of evolution
Structure, 17 (5), pp. 637-650
Abstract | PDF Article
2008

Bart Van Oort, Alexey Amunts, Jan Willem Borst, Arie Van Hoek, Nathan Nelson, Herbert Van Amerongen, Roberta Croce
Picosecond fluorescence of intact and dissolved PSI-LHCI crystals
Biophysical Journal, 95 (12), pp. 5851-5861
Abstract | PDF Article

Alexey Amunts, Nathan Nelson
Functional organization of a plant Photosystem I: evolution of a highly efficient photochemical machine
Plant Physiology and Biochemistry, 46 (3), pp. 228-237
Abstract | PDF Article
2007

Alexey Amunts, Omri Drory, Nathan Nelson
The structure of a plant photosystem I supercomplex at 3.4 Å resolution
Nature, 447 (7140), pp. 58-63
Abstract | PDF Article
2005

Alexey Amunts, Adam Ben-Shem, Nathan Nelson
Solving the structure of plant photosystem I—biochemistry is vital
Photochemical & Photobiological Sciences, 4 (12), pp. 1011-1015
Abstract | PDF Article